United Kingdom
United Kingdom Argentina
Argentina  Australia
Australia  Austria
Austria  Brazil
Brazil  Canada
Canada  Germany
Germany  Ireland
Ireland  Italy
Italy  Malaysia
Malaysia  Mexico
Mexico  New Zealand
New Zealand  Poland
Poland  South Africa
South Africa  United States
United States Bacterial N4-methylcytosine as an epigenetic mark in eukaryotic DNA

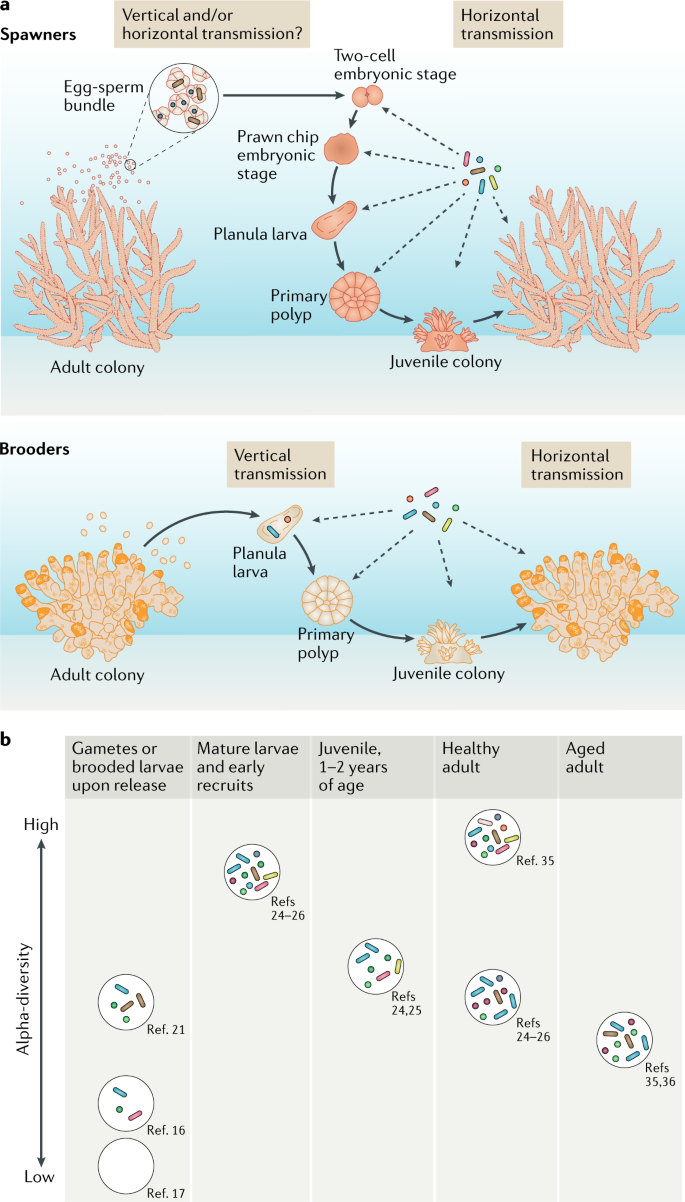
A bacterial amino-MTase in bdelloid rotifers
Rotifers of the class Bdelloidea are tiny freshwater invertebrates a fraction of a millimeter long, characterized by clonal reproduction, eutely, direct development, syncytial tissues, and paleotetraploid genome structure15. They are known for an unmatched ability to incorporate foreign genes into genomic DNA, largely preserving their functionality16. In sequenced bdelloids, 8–12% of coding sequences are of non-metazoan, mostly bacterial, origin17,18,19. Surprisingly, we found that one such bacterial gene in the sequenced bdelloid Adineta vaga17 is represented by an allelic pair of MTases containing the N6_N4_MTase domain (PF01555), which is closely related to amino-MTases of bacterial R–M systems acting on the exocyclic amino group of adenines and cytosines (Fig. 1a; Supplementary Fig. 1). Its orthologs, sharing the same five conserved intron positions, are present in sequenced representatives of each major family of the class Bdelloidea, dating back 40–60 Mya, but are absent from sequenced members of the sister class Monogononta or from other sequenced eukaryotes (Fig. 1e, f; Supplementary Fig. 2). Both classes, however, encode amino-MTases implicated by various authors in adding 6mA marks to eukaryotic DNA: METTL4-like (PF05063: MT-A70), N6AMT1-like (PF05175: MTS) and N6AMT2-like (PF10237: N6-adenineMIase)12,20,21,22,23 (Fig. 1b, f). Notably, none of the sequenced rotifers harbor Dnmt1 or Dnmt3 genes for the most common eukaryotic C5-MTases, encoding only the tRNA-modifying Dnmt2/Trdmt.

a, b Domain structure of putative N4C (a) and N6A (b) bdelloid amino-MTases. PFAM/KOG domains are indicated; conserved catalytic motifs and S-adenosylmethionine (AdoMet) binding sites are flagged; numbers correspond to aa positions in A. vaga. See Supplementary Data 1 for gene IDs and aa sequences. c, Immuno-dot-blot analysis of membrane-immobilized gDNA from A. vaga Av-ref (746 ng), AvL1 (500 ng), E. coli C2925 dam−/dcm− (550 ng) and E. coli M28 (2 µg) probed with anti-4mC (top panel) and anti-6mA (bottom panel) antibodies. d, Summary of gDNA digestion (+) with restriction enzymes differing by methylation sensitivity: MboI (blocked by dam methylation); DpnI (cleaves only dam methylated DNA); Sau3AI (not sensitive to dam or dcm methylation); McrBC (cleaves at any methylated cytosine). e Neighbor-joining phylogram of permuted MTases of Type II, subtype β, displaying clustering by recognition sequences obtained from REBASE. Clustering is not intended to uncover phylogenetic relationships in bacteria. Red arrow indicates acquisition of a chromodomain (CHD) by the bdelloid N4CMT. Sequences are provided in Supplementary Data 1. f Phyletic distribution patterns of putative DNA methyltransferases implicated in 4mC, 6mA, and 5mC addition. A consensus cladogram of metazoan phyla is shown on the left. g Adineta vaga AvL1 under polychromatic polarizing microscope. Photo credit: M. Shribak, I. Yushenova. Scale bar, 50 µm.
The N6_N4_MTase found in A. vaga belongs to the permuted type, in which the catalytic domain is located N-terminally to the S-adenosylmethionine (AdoMet) binding domain24 (Fig. 1a). Its evolutionary history and taxonomic distribution (Fig. 1e, f) differs dramatically from that of 5mC- or N6A-MTases6. The small non-permuted pan-eukaryotic MTases N6AMT1 and N6AMT2 (Fig. 1b), variably annotated either as N(6)-adenine MTases or as small N5-glutamine (HemK-like) and lysine (eEF1A) MTases, respectively, have been implicated in N6A methylation based on knockout/knockdown data21,25, but do not carry N- or C-terminal extensions, and modify proteins rather than DNA in functional assays26,27,28,29,30, suggesting that in vivo perturbations may have indirect effects. The presumptive N6A-MTase METTL4, which in Drosophila adds m6A to U2 snRNA in vitro31, has a conserved N-terminal domain (KOG2356: transcriptional activator, adenine-specific DNA methyltransferase) present in METTL4-like ORFs of most eukaryotes, including A. vaga (Fig. 1b, top). This permuted MTase, found in all bdelloids, may have persisted in eukaryotes throughout their evolutionary history (Fig. 1f, Supplementary Table 1). In contrast, the bdelloid N6_N4_MTase has no eukaryotic homologs, and can be aligned only with permuted bacterial N4C- and N6A-MTases (Type II, subtype β), which cluster in accordance with their target recognition domains (TRD) recognizing specific targets compiled in REBASE1,24,32 (Fig. 1e; Supplementary Data 1). Interestingly, the bdelloid lineage clusters with phage MTases of unknown target specificity, and its closest bacterial homologs are N4C-MTases recognizing TCGA and CCSGG. Thus, we tentatively assigned it to N4C-MTases and named it N4CMT, since it harbors the catalytic SPPY motif shared with most bacterial N4C-MTases and differing from bacterial N6A-MTases (DPPY), eukaryotic N6AMT1 (NPPY), N6AMT2 (DPPY/F) or METTL4-like enzymes (DPPW, also seen in METTL3/IME4-like m6A-RNA MTases)12,24,33 (Supplementary Table 2).
Presence of 4mC and 6mA marks in genomic DNAWe next sought to find out whether recruitment of a horizontally transferred bacterial MTase resulted in the establishment of bacterial epigenetic marks in bdelloid genomic DNA (gDNA). A strong indication that N4CMT could interact with chromatin to add 4mC to gDNA comes from the presence of a eukaryotic chromodomain from the HP1/chromobox subfamily of methylated lysine-binding Royal family of structural folds34 at the C-terminus of the bacterial N6_N4_MTase moiety in sequenced bdelloids (Fig. 1a, Supplementary Fig. 2).
To detect 4mC/6mA marks in bdelloid genomes, we extracted gDNA from the A. vaga laboratory reference strain (hereafter Av-ref)17 fed with methyl-free Escherichia coli (Supplementary Table 3), and performed immuno-dot-blotting with anti-4mC and anti-6mA antibodies (Methods). We also extracted gDNA from the natural A. vaga isolate L1 (hereafter AvL1; Supplementary Movie; Fig. 1g), which was caught in the wild and identified as A. vaga through morphological criteria and mtDNA phylogeny, but represents a distinct morphospecies within the A. vaga species complex, as its gDNA is only 88% identical to Av-ref35. Figure 1c shows that gDNA from Av-ref and AvL1 reacts positively with both antibodies, suggesting the presence of 4mC and 6mA marks. Control DNAs isolated from the dam-/dcm-, DH5α and Top10 E. coli strains, or from E. coli M28 strain used as food (Supplementary Table 3), did not react with anti-4mC antibodies (Fig. 1c), and neither we observe cross-reactivity of the anti-4mC antibody with 5mC-containing human DNA. Also consistent with the presence of modified cytosines were the results of treatment of total A. vaga gDNA with the McrBC endonuclease, which cleaves at any methylated cytosines (5mC, 5hmC, 4mC)36,37 (Fig. 1d; see also Fig. 3b below). Together with the absence of C5-MTases, the similarity of N4CMT to bacterial N4C-MTases (Fig. 1e), and the lack of 5mC deamination signatures in gDNA from observed/expected CpG ratios (Supplementary Fig. 3a), our data support the hypothesis that cytosines in bdelloids are modified at the N4- rather than C5-positions. Still, signals in gDNA may originate from residual methylated bacterial DNA from sources other than food. Thus, we sought to examine the distribution of 4mC marks over annotated genomic features in bona fide eukaryotic contigs.
Genome-wide analysis of 4mC and 6mA by DIP-seqWe exploited immunoreactivity of bdelloid DNA with anti-4mC and anti-6mA antibodies to assess the genome-wide distribution of these methylation marks by DIP-seq (DNA immunoprecipitation followed by sequencing, also called MeDIP-seq; see Methods). After read mapping to Av-ref, MACS peak-calling tool identified 1008 and 1735 DIP-seq peaks (p-value 2a, left and right), suggesting that TE insertions could be an important 4mC modification target. For gene annotations, IP occupancy (Fig. 2a, center) does not show an increase in density at the transcription start site (TSS) seen in TEs. After peak calling with MACS, we also compared relative numbers of peaks with intersected annotations: about one-half of 4mC peaks (468 out of 1008) and a quarter of 6mA peaks (430 out of 1735) are close to TEs, and more 6mA peaks (1261 out of 1735) than 4mC peaks (398 out of 1008) are close to gene annotations. Genometric spatial correlation analysis (Supplementary Note 1; Supplementary Table 4; Supplementary Fig. 4) further shows that DIP-seq 4mC marks are closer to TEs than would be expected from a uniform distribution.

a Distribution of DIP-seq 4mC and 6mA sites around TEs (metaprofile), genes (TSS), and 5′-end TE profiles in Av-ref, showing IP occupancy in 25-bp bins within ±2.5 kb of each feature. In metaprofiles, the body size feature, representing genes or TEs, is automated and normalized (0–100% length). TE 5′-profile shows 4mC and 6mA sites near 5′ boundaries, aligning transposons at the 5′ end. b IPD ratios in AvL1 SMRT-seq data at four representative 4mC and 6mA modification sites. Purple and orange bars, Watson and Crick strands. c SMRT-seq 4mC and 6mA occupancy in 5 and 25-bp bin sizes within ±0.5 and ±2.5 kb of 5′ TE boundaries. d MEME-ChIP motif analysis of regions around SMRT-seq 4mC and 6mA sites. Windows of ±5, ±10, and ±20 bp were extracted and searched for significant motif enrichment. Significance was assessed by Fisher’s exact test; p-value generated by MEME-ChIP is shown under each motif. e Methylation fraction distribution at modified sites detected by SMRT-seq. Most 4mC sites are fully methylated (fraction = 1); average methylation level of 6mA sites is 0.74. f PacBio read coverage distribution by base modification sites. The minimal threshold coverage limit applied for calling 4mC and 6mA methylated sites to calculate methylation fraction per site in (e) is shown by a dashed line. g Average numbers of 4mC and 6mA base modifications in protein-coding genes, TEs, and tandem repeats. Average is calculated as the total number of modified sites divided by the total number of annotations (unique IDs) in each feature and divided (normalized) by the genome fraction covered by such annotation in the genome (genes, 0.533; TE, 0.021; TR, 0.0084). h Distribution of SMRT-seq 4mC and 6mA sites within genic features (CDS, intron, 5′ UTR, 3′ UTR, 5′-promoter region) and intergenic regions by average feature size (bp). i DNA methylation density vs. TE copy integrity. Bar height indicates average number of 4mC or 6mA SMRT counts; error bars represent standard deviation for full (n = 321), medium (n = 305), and short (n = 8623) TE copies.
The presence and distribution of 4mC and 6mA DNA modifications in the AvL1 strain were similarly interrogated by DIP-seq. We generated DIP-seq reads and mapped them onto AvL1 assembly (Methods). After peak calling with MACS, we identified 1473 and 1385 peaks (p-value 5). AvL1 repeat library was constructed de novo, manually curated, and used to annotate TEs (Methods). Initial analysis showed that 4mC-DIP-seq and 6mA-DIP-seq have similar distribution profiles in the assembly (Supplementary Fig. 5a, b) with enrichment of both marks towards genes and transposons, part of which may be due to the undetected presence of unknown types of low copy-number TEs in gene annotations. Nevertheless, cluster analysis showed an increase in 4mC being detected in a subset of transposons (clusters 1 and 2, Supplementary Fig. 5d). After peak calling, we found 1097 4mC peaks (out of 1473) and 1042 6mA peaks (out of 1385) close to TEs, while 863 4mC and 813 6mA peaks are close to genes (excluding TEs). Genometric correlation analysis in AvL1 showed that both 4mC and 6mA modification peaks (Supplementary Note 4; Supplementary Table 4; Supplementary Fig. 4) display a small absolute positive correlation with TEs, being closer than expected to TEs than to gene models as reference features (Jaccard and permutation test). Together, DIP-seq data in both Av-ref and Av-L1 suggest preferential localization of 4mC over TEs.
Modification analysis at single-base resolution by SMRT-seqWhile immuno-dot-blots and differential gDNA digestion suggested the presence of 4mC in bdelloid gDNA, one cannot fully eliminate gDNA from commensal bacteria, even using methyl-free E. coli food strains and applying starvation/antibiotic treatments prior to DNA extraction (Methods). Hence, we chose not to use mass-spectrometry (MS) as a method to confirm the presence of 4mC in bdelloids, especially considering that unknown MS-peaks can comigrate with 4mC14. Further, the low resolution of the DIP-seq method limits the power of correlation analyses to the length of DNA fragments used for antibody binding (250–450 bp), not to mention residual IgG binding to non-modified fragments inherent to the method38. Thus, we chose to examine the genome-wide distribution of modified bases by single-molecule real-time (SMRT) sequencing, which provides single-nucleotide resolution and allows validation of metazoan/bacterial contigs (Methods).
SMRT-based detection exploits kinetic signatures of polymerase passage through modified vs non-modified bases and is quantified in terms of inter-pulse duration (IPD) ratios. It is best suited for the detection of 4mC and 6mA, characterized by strong kinetic signatures, which require ~10-fold lower coverage than 5mC detection (Pacific Biosciences Methylome Analysis Technical Note) and is widely used in bacterial methylome analyses32,39. We obtained PacBio reads (15 SMRT cells, totaling 9.87 Gb) from gDNA extracted from AvL1 eggs and analyzed the kinetic profiles with SMRT® Portal (Methods). Prior to quantification of modified bases, we bioinformatically removed residual bacterial contigs, which show high methylation density.
SMRT-analysis detected 4mC modifications on 21,016 cytosines (0.0643% of the total cytosines in the assembly) and 6mA modifications on 17,886 adenines (0.0236% of total adenines) using a minimum cutoff PacBio coverage defined in Fig. 2f (see Supplementary Table 6 for comparison of 10× and 20× coverage levels). As with DIP-seq, SMRT-seq shows a broad distribution of both modifications across AvL1 assembly. Comparison of DIP-seq and SMRT-seq modification patterns shows considerable overlap, with 36% of 4mC peaks and 32% of 6mA peaks overlapping with 4mC and 6mA identified by SMRT analysis, respectively, indicating that many peaks are conserved between eggs and adults. Following normalization of SMRT-seq methylation fraction values per modified base (see Methods), it is seen that 4mC and 6mA DIP-seq peak summits overlap with modified regions for PacBio 4mC and 6mA, respectively; plotting only 4mC-marked CpG sites shows a similar increase towards DIP-seq 4mC peaks (Supplementary Fig. 6a). The peak overlap is quite substantial, given the modest proportion of modified bases in the genome, and might reflect the general lack of methylation reprogramming during development in protostomes, known at least for 5mC40.
In contrast to the predominantly symmetric patterns of 5mC deposition at CpG doublets in eukaryotes, AvL1 shows mostly asymmetric patterns of methylation for both 4mC and 6mA, i.e., only one strand is usually modified (Fig. 2b displays typical examples). At 4mC sites, CpG and CpA dinucleotides are the most prevalent, making up 74% of modified doublets. For better identification of sequence preferences, we extracted different sequence windows (5, 10, and 20 bp) upstream and downstream from 4mC sites and searched for significant motif enrichment with MEME-ChIP (Methods) (Fig. 2d). For 4mC, three motifs with CG or CA dinucleotides were most significantly enriched (from p = 2.8e−593 to p = 1.4e−513). For 6mA, a similar approach yielded three significantly enriched short motifs (from p = 7.3e−656 to p = 4.3e−420); increasing the motif length yielded GA embedded in an A-rich region (p = 2.4e−1243). However, none of these matched the RRACH motif found at m6A sites in RNA41, arguing against RNA contamination. The dinucleotide GA is the most prevalent at 6mA sites, and the most common triplets AGG or GAA, when combined, compose 34% of all 6mA triplets. These findings parallel the known 6mA motif preferences in metazoans but differ from unicellular eukaryotes and early diverging fungi, in which 6mA methylation is symmetric and targets ApT dinucleotides (Supplementary Table 1).
In addition to measuring coverage at each 4mC and 6mA site, the SMRT-analysis pipeline reports different methylation levels (fraction), referring to the proportion of times a given nucleotide is identified as methylated (1 equals 100% methylation). Notably, most 4mC methylation corresponds to high-fraction sites (0.5–1), dominating over low-fraction sites (0.1–0.5) at a ratio 71:1, with 58% of 4mC sites being fully methylated (Fig. 2e). Methylation at 6mA sites appears more dynamic, although the highly methylated (0.8–1) and moderately methylated (0.5–0.8) sites still dominate over low-fraction sites (0.1–0.5), which constitute only 12% of 6mA sites.
We plotted the density of 4mC and 6mA in AvL1 (DIP-seq and SMRT-seq) across annotated features (genes, TEs, tandem repeats (TRs)) (Fig. 2c, g; Supplementary Fig. 5a–d). The 4mC and 6mA tag densities are close for each annotation type (Fig. 2g). The 4mC density in SMRT-seq is higher near TE 5′-ends (Fig. 2c), as was also seen in Av-ref DIP-seq showing increased deposition of 4mC peaks close to TE 5′ ends (Fig. 2a, right). Enrichment of 4mC over TEs, especially near 5′-boundaries, was also preserved for fractional methylation values in SMRT-seq metaprofiles (Supplementary Fig. 6b). Nevertheless, a fair number of 6mA sites (DIP-seq and SMRT-seq) is found near TEs (Fig. 2g, i; Supplementary Fig. 5b, d).
Methylation density in TRs deserves special mention. Figure 2g shows that the average counts of 4mC and 6mA sites in TRs are elevated in comparison with TEs and genes. According to TR annotation (Methods), only a small fraction (0.84%) of the AvL1 assembly is composed of TRs. Inspection of SMRT-seq modification data identified two repeats with a very high density of methylated sites, located mainly on contigs 1882 and 785 adjacent to large Athena retroelements42. Such extra-high modification density, approaching that in bacterial contigs, mostly accounts for over-representation of modified bases in TRs, leaving other TRs virtually unmethylated. In subsequent experiments, we took advantage of the high methylation susceptibility of these repeats (see below).
In genes, the PacBio methylation tag density is much lower than that in TEs and TRs (Fig. 2g). Still, genic regions cover slightly over one-half of the AvL1 genome, attracting a sizeable proportion of 4mC and 6mA modifications (52% of 4mC and 54% of 6mA). To correlate methyl marks with gene structure, we examined 4mC and 6mA distribution using more refined features: gene bodies, promoters within 2 kb upstream of the TSS, and intergenic regions which may include TEs and TRs, with gene bodies further subdivided into CDS (exons excluding 5′ and 3′ UTRs), introns, 5′ and 3′ UTRs (Fig. 2h). Altogether, base modifications are found in all features (CDS, promoters, and intergenic regions); when the density per average feature size is compared, CDS regions carry more 4mC than introns (Fig. 2h), reminiscent of 5mC patterns in mammals43, but introns carry as many 6mA marks as CDS, minimizing the possibility of m6A carryover from RNA.
In AvL1, DIP-seq shows relative enrichment with 4mC and 6mA within TE bodies (Supplementary Fig. 5b, d). PacBio 4mC sites display a trend for enrichment near the 5′ TE boundaries, while 6mA sites show a local depletion (Fig. 2c), which is visible even though TE promoters are located near TE 5′-ends but not necessarily at the boundary, and is not due to a local change in base or dinucleotide composition (Supplementary Fig. 3b). Moreover, 4mC and 6mA marks are predominantly found over full-length or nearly full-length TE copies and are practically absent from shorter TE fragments spanning less than one-half of TE consensus length, suggesting that active TE copies are preferentially targeted (Fig. 2i). The lack of 4mC and 6mA marks in shorter TE copies, together with a concentration of 4mC near 5′ TE boundaries, suggest that their deposition is associated with transcriptional activity.
To visualize 4mC and 6mA densities in TRs, TEs, and genes on representative contigs, we built Circos plots (Supplementary Fig. 7a–d), in which the PacBio modification layer is plotted as modification fraction (from 0 to 1) for each modified base. In agreement with Fig. 2e, highly methylated 4mC sites dominate in most locations, while 6mA sites are distributed over a much wider methylated fraction range and across a wider feature range. Importantly, higher densities of modified bases are not necessarily correlated with areas of higher PacBio read coverage, indicating that over-representation of methyl marks over TEs and TRs is not due to excess coverage in these regions (e.g. mtDNA at 127x coverage displays very few marks) (Supplementary Fig. 7e). Supplementary Fig. 7c, d shows that long copies of Vesta and Athena retrotransposons attract methyl marks, but short copies do not. Supplementary Fig. 8 presents a more detailed view of selected contigs, including TRs, retroelements, and DNA TEs. Of note, an inspection of 36 high-density 4mC regions lacking annotations showed that one-half correspond to TEs unrecognized during annotation, independently confirming TEs as N4CMT targets (Supplementary Fig. 7f; Supplementary Table 11; Supplementary Note 3).
N4CMT acts as 4mC-methyltransferase in E. coliThe domain structure of N4CMT cannot be taken as evidence of its N4C-MTase activity, since the N6_N4_MTase domain repeatedly evolved 6mA or 4mC specificities44. However, N4CMT function cannot be disrupted in vivo, as the tools for genetic manipulation in bdelloids are yet to be developed. We, therefore, sought to investigate the activity of the recombinant N4CMT protein in a heterologous system. To this end, we PCR-amplified N4CMT from A. vaga cDNA to obtain intronless versions (Methods; Supplementary Table 7). Amplicons were cloned into pET29b expression vector with the N-terminal S-tag and the C-terminal 6×His-tag and expressed in E. coli. We examined two A. vaga allozymes A and B, differing by six amino acids (aa): three in the N6_N4_MTase domain and three in the chromodomain-containing C-terminus (Supplementary Table 8; Supplementary Fig. 9a). We also tested two inter-allelic recombinants swapping the rightmost substitution near the C-terminal His-tag, which may have arisen during rotifer cultivation or PCR amplification, as well as two 3′-truncated derivatives lacking the chromodomain.
To assess plasmid-borne N4CMT activity in vivo, its expression was induced by IPTG, and gDNA was extracted 4 h post-induction (Methods). Figure 3a shows the immuno-dot-blot of membrane-immobilized gDNAs probed with anti-4mC and anti-6mA antibodies, with 4mC signal observed from full-length N4CMT allozymes in the absence of signal from the untransformed host strain. As expected in the dam + background, 6mA methylation was detected in all samples, serving as an internal DNA control. Not surprisingly, removal of the chromodomain, which yields a core MTase equal in length to its bacterial counterparts, did not reduce activity and even showed an increase in signal intensity due to better solubility of the 33- vs. 45-kDa enzyme (Fig. 3a, N4CMT-ΔCbx). The N4CMT_A allozyme mostly showed weaker activity, suggesting that substitutions in the presumed TRD region of the N6_N4_MTase domain affect protein solubility or interaction with target DNA. These findings were corroborated by digestion of corresponding gDNAs with the endonuclease McrBC, which cleaves DNA at any modified cytosines. DNAs extracted from Rosetta 2(DE3) transformed with six N4CMT-expressing plasmids and the control human DNA were readily digested with McrBC, while DNA from the untransformed dcm- strain was not (Fig. 3b).

a Immuno-dot blot of total DNA extracted from E. coli Rosetta 2(DE3) strain transformed with different variants of recombinant N4CMT. DNA from non-transformed Rosetta 2(DE3) was used as a control. DNA was extracted after 4 h of IPTG-induced N4CMT expression; 400 ng of each DNA was spotted in one dot. Methyl marks are indicated on the right. b Methylcytosine-sensitive digestion of E. coli gDNA. Total DNA from Rosetta 2(DE3) strain, either transformed with recombinant N4CMTs (as shown on the top) or untransformed, was extracted 4 h post-induction; DNA (6.3 μg) was treated with McrBC. DNA from HepG2 liver cell line (H. sapiens) was used as a positive control (500 ng). Two independent experiments were performed with similar results. c Immuno-dot blot of total E. coli DNA showing the role of the SPPY motif in N4CMT methylation. Same designations as in (a). d, Immuno-dot blot of total E. coli DNA extracted from different strains and treated with N4CMT. Rosetta 2(DE3) and dam−/dcm−, 950 ng per dot; BL21-AI and M28, 500 ng per dot. e Immuno-dot blot with anti-4mC antibody for sAvL1-451 substrate treated with N4CMT allozymes and their catalytic site mutants in vitro.
To ensure that the observed activity is directly attributable to N4CMT, we created N4CMT mutants in which the catalytic SPPY motif was replaced with APPA (Supplementary Table 8). Figure 3c shows that 4mC addition is abolished after substitution of the catalytic Ser and Tyr residues with Ala, indicating that N4CMT is responsible for adding N4-methyl groups to cytosines in dsDNA with SPPY as the catalytic motif, justifying our initial N4CMT designation. Further investigation of purified recombinant N4CMT activity on preferred substrates in vitro revealed that it acts de novo on unmethylated dsDNA, and that a conserved sequence motif in the TR mediates sequence-specific mode of substrate recognition (Supplementary Note 2; Supplementary Fig. 9).
Base modifications and histone modificationsIn the context of eukaryotic chromosomal DNA environment, any intrinsic target preferences of N4CMT manifested in vitro, while apparently yielding high 4mC densities in certain TRs, would not necessarily be required for 4mC deposition in other genomic regions, which may instead be facilitated by the C-terminal chromodomain of the chromobox type (CBX)34. CBX is expected to recognize methylated lysines K9 and K27, the best-studied heterochromatic marks embedded in the ARKS motif at the N-terminus of histone H3, which are associated with transcriptionally silent chromatin and in non-mammalian systems frequently overlap, not because of antibody cross-reactivity but due to a similar function in TE repression45,46,47,48. To associate DNA methylation marks with specific histone modifications, we performed chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) on A. vaga chromatin with anti-H3K9me3 and anti-H3K27me3 antibodies (Methods). For contrasting comparisons with active chromatin, we used an anti-H3K4me3 antibody, which recognizes the modification associated with active TSS45,49. After validating antibodies by immuno-dot-blotting (Methods), we profiled the distribution of three H3K modifications in Av-ref and AvL1 strains by ChIP-seq. We found that H3K9me3, a mark for constitutive heterochromatin, often co-localizes with H3K27me3 known to characterize facultative heterochromatin, but not with H3K4me3 which marks active genes (Supplementary Table 10). As expected, host genes display significant H3K4me3 enrichment, which typically covers 1–2 kb around the TSS and shows a characteristic bimodal peak in both strains (Fig. 4a, c top). In contrast, H3K9me3 and H3K27me3 enrichment is observed mostly over TEs and covers the entire TE body, often extending upstream and downstream from a TE insertion, which may be indicative of spreading (Fig. 4b, d top).

Shown are the A. vaga strains Av-ref (a, b, e) and AvL1 (c, d, f). a, c Profiles and heatmaps for gene regions with transcription start sites (TSS) and transcription termination sites (TTS). Profiles (top) show mean ChIP-seq signal for H3K4me3, H3K9me3, and H3K27me3. Heatmaps (bottom) represent H3K4me3, H3K9me3, and H3K27me3 ChIP-seq reads, where each row corresponds to gene regions with ±3 kb from TSS and TTS boundaries. Heatmap color bars represent H3K enrichment normalized by RPGC (reads per genome coverage). b, d Profiles and heatmaps for TE annotations delimited by 5′ and 3′ boundaries. Profiles (top) show mean ChIP-seq signal for H3K4me3, H3K9me3, and H3K27me3. Heatmaps (bottom) represent H3K4me3, H3K9me3, and H3K27me3 ChIP-seq reads, where each row corresponds to ±3 kb from 5′ or 3′ TE boundary. e, f Intersection of 4mC and 6mA DIP-seq peak summits with ChIP-seq for histone modification marks H3K4me3, H3K9me3, and H3K27me3 for Av-ref (e) and AvL1 (f). The log2 ratio is shown over a scaled window ±3 kb. y-Axis, relative fold enrichment. g DNA base methylation counts near H3K4me3, H3K9me3, and H3K27me3 ChIP-seq peaks. Counts are taken around each peak in a ±500 bp window. The bar height shows an average number of counts (SMRT-seq 4mC and 6mA). Error bars represent standard deviation for H3K4 (n = 5789), H3K9 (n = 1205), and H3K27 (n = 2378) peaks. h Circos plot illustrating DIP/ChIP peaks, methylation sites, sequencing read coverage, and gene/TE annotations in selected Av-ref and AvL1 contigs. Features are explained in the key; further details are in Methods and Supplementary Fig. 7. SMRT-seq DNA methylation marks are shown within the PacBio layer in AvL1 for 4mC (blue triangle) and 6mA (red square). Mark height in the ring shows a methylation fraction (0–1). Green line, RNA-seq coverage; purple line, small RNA coverage in Av-ref.
To explore the association of 4mC and 6mA with active or repressive histone marks, we used ChIP-seq data for the euchromatic mark (H3K4me3) and two heterochromatic marks (H3K9me3 and H3K27me3) as a proxy for active and silent chromatin, respectively. The low resolution of DIP-seq precludes genome-wide extrapolations in Av-ref, allowing only initial comparisons. For 6mA-DIP-seq peaks, 13.6% intersected with regions bearing euchromatic histone modifications (H3K4me3), while only 4.4% overlapped with heterochromatic histone modifications (H3K9me3 and H3K27me3 combined). For 4mC-DIP-seq peaks, 6.5% intersected with regions bearing heterochromatic histone modifications (H3K9me3 and H3K27me3), but only a minor fraction (1.5%) overlapped with H3K4-marked regions. Following normalization and aggregation of aligned reads in ChIP-seq datasets and comparison with chromatin DNA input (log2 ratio with bamCompare; see “Methods”), we found that DIP-seq peak summits (4mC and 6mA) overlap with H3K9me3 and H3K27me3 ChIP-seq covered regions, while little if any overlap is seen with H3K4me3 (Fig. 4e).
In AvL1, for 4mC-DIP-seq peaks, 42.3% intersected with regions bearing heterochromatic histone modifications (H3K9me3 and H3K27me3 combined), but only 6.6% overlapped with H3K4me3-marked regions. Similarly, for 6mA-DIP-seq peaks, 42.9% overlapped with heterochromatic histone modifications (H3K9me3 and H3K27me3 combined), but only 6.3% intersected with regions bearing euchromatic H3K4me3 modifications. After normalization of aligned reads in the ChIP-seq dataset and comparison with chromatin DNA input, we confirmed that DIP-seq peak summits (4mC and 6mA) are strongly correlated with H3K9me3 and H3K27me3 heterochromatic ChIP-seq reads, as seen in Fig. 4f. Examples of co-localization may be seen in Fig. 4h and Supplementary Fig. 8. Thus, the presence of DNA methyl marks is preferentially associated with silent chromatin in both strains. A similar pattern is observed in AvL1 SMRT analysis, where the 4mC and 6mA marks are more frequently associated with inactive chromatin domains marked by H3K9me3 and especially H3K27me3 (Fig. 4g). Collectively, these results support the view that, in addition to any intrinsic target preferences of N4CMT, its action in the genome may be directed by the CBX moiety, targeting MTase activity to chromatin regions with repressive histone marks.
Methylomes and transcriptomes in the chromatin contextTo associate histone marks with transcriptionally active or repressed genes in A. vaga, we plotted our RNA-seq data for genes co-localizing with either active or repressive H3K-me3 histone marks (“Methods”). As expected, genes near H3K4me3 have significantly higher RPKM (reads per kilobase of transcript per million mapped reads) values (ANOVA p-val 5a). AvL1 displays the same pattern (Supplementary Fig. 10a). The tentative designation of 6mA modification as an active epigenetic mark9,13 prompted us to similarly explore its correlation with gene transcription. The A. vaga gene dataset, after removing TE-derived genes, was divided into two groups, with and without the presence of 6mA peaks within a window size of ±500 bp of each gene ID, and RPKM values were counted in both groups. We found that genes with 6mA depositions tend to have higher RPKM than genes without 6mA (t-test p-val: 2.2E−16, Fig. 5b bottom). For genes with 4mC modifications, no significant differences in expression were seen with or without 4mC marks (Fig. 5b top). A detailed analysis of 6mA distribution in genes and their promoters, which shows that only a subset of genes is affected, and rules out contribution of m6A from RNA, is presented in Supplementary Note 3 and Supplementary Figs. 11 and 12 (see also Source Data 1).

a Box plot showing Av-ref gene expression levels (log2RPKM) associated with co-localized H3K4me3, H3K9me3, H3K27me3, and both H3K9-27me3 marks or without histone marks. ANOVA analysis (one-sided with Tukey’s post hoc; p-val b, c RPKM values associated with DIP-seq 4mC and 6mA base modifications in A. vaga. TE genes in (c) are derived from Av-ref automated gene models after positively intersecting with TE annotations. Box plots indicate the first and third quartiles; line, the median; single points as outliers. Error bars are calculated as the standard deviation of three biological replicates. The p-values were calculated by a two-tailed Student’s t-test, with asterisks indicating significant differences.
A different picture was observed for TEs upon examining the association of transcript levels of TE-related genes with DIP-seq peaks (“Methods”). While TE-related genes with or without 6mA did not show much difference in RPKM values, TE-related genes with 4mC marks showed a decrease when compared to those without 4mC (t-test p-val: 6.8E−8, Fig. 5c, top). Thus, in expressed TEs 4mC may be regarded as a repressive mark. Note that co-localization of 4mC and 6mA is compatible with repression, as 6mA was reported to form an adversarial network preserving Polycomb silencing23. Alternatively, some of the 6mA marks co-localizing with 4mC-rich regions may represent a “bleed-through” signal from the nearby 4mC in SMRT-seq data, as was inferred for 5mC-rich regions in mammals11. Regardless of 6mA role, the transcriptionally repressed state of TEs is corroborated by a measurable overlap with small RNA profiles, observed for 4mC but not for 6mA (Supplementary Note 4; Supplementary Fig. 13c). Small RNAs play a prominent role in transcriptional repression of A. vaga TEs50, and bdelloids show a dramatic expansion of RNA-mediated silencing machinery, with dozens of Piwi/Ago and RdRP copies17,51.
Interpreting the 4mC marksTo identify possible readers of bacterial marks, we searched for candidate proteins capable of discriminating between methylated and unmethylated cytosines. All known DNA methyl groups protrude from the major groove of the B-form double helix and can be recognized as epigenetic marks. In eukaryotes, several protein domains can read 5mC (SRA/SAD/YDG; MBD/TAM; Kaiso) or 6mA (HARE-HTH; RAMA)6,12, usually in a preferred sequence context. We used profile-HMM searches to find candidate methyl readers in Adineta genomes. No homologs were found for the SAD_SRA domain (PF02182), which recognizes hemimethylated CpGs by embracing DNA and flipping out the methylated cytosine52. However, we saw the drastic expansion of MBD/TAM-containing proteins, which do not require base-flipping: 14 different alleles (originating from three quartets, Q1–Q3, plus a segmental duplication) encode seven SETDB1 variants, as opposed to only one in monogonont rotifers or other invertebrates (Fig. 6a, b; Supplementary Fig. 14a; Supplementary Data 2). These proteins share the same domain architecture, with the MBD sandwiched between the N-terminal triple-Tudor domains and the C-terminal pre-SET/SET/post-SET domains, present in all SETDB1/eggless-like H3K9me3 histone lysine MTases (KMTs) (Fig. 6a). All seven proteins are transcribed in each Adineta spp. (Supplementary Fig. 15). Additional MBD/TAM domains of BAZ2A/TIP5-like remodelers, which form heterochromatin on rDNA and satellites53, comprise only one quartet in A. vaga (Supplementary Fig. 14c; Supplementary Data 3).

a Domain architecture of bdelloid SETDB1 proteins. Square bracket marks cloned MBD domains; aa numbering is for Av_s314. b Unrooted maximum likelihood phylogram of SETDB1 variants in bdelloids (blue), monogononts (green), and acanthocephalan (olive). Q1–Q3, quartets of homeologs formed by paleotetraploidy. Bottom clades include single-copy SETDB1 in 3 protostome phyla. See Supplementary Data 2 for aa sequences. Scale bar, aa substitutions per site. c LINE retrotransposon content (% genome) and Piwi/Ago copy numbers in 6 monogonont (green) and 10 bdelloid (blue) species (Supplementary Table 2; Source Data 2). Median values are indicated by bar heights for LINE content, and by continuous orange line for copy numbers. Data points for % LINE values (+) and Piwi/Ago copy numbers (♦) are shown for all sequenced isolates from each species, with number of isolates (n) in parentheses. d Affinity of AvMBD for 4mC-methylated DNA in electrophoretic mobility shift assays. EMSA was performed using 0.05 nM 32P-labeled sAvL1-451 DNA and 3.75 nM AvMBD_s314 protein. Unmethylated and 4mC-methylated by N4CMT_B sAvL1-451 fragments were used as competitor DNA. One of two gels with the same DNA:protein ratio from independent experiments is shown. e AvMBD_s314 DNA binding preference for methylated DNA. Y-axis, percent unbound 32P-labeled sAvL1-451 DNA for 3.75 nM AvMBD_s314 in the presence of unmethylated and 4mC-methylated sAvL1-451 competitor DNA. Median values are indicated by continuous lines, with dots representing the data points for two independent experiments. Five independent experiments, including those with different DNA:protein ratios, are presented in Source Data 2. f A hypothetical model of the self-reinforcing regulatory loop based on the ability of N4CMT and SETDB1 to cross-recognize methyl marks on histones (circle) and DNA (square), respectively. Shown are the relevant conserved proteins/domains described in the text. Shadows, multiple copies. Hypothesized links with piRNAs/Piwi (dashed lines) are not defined. [X] and [?] are hypothetical mediator complexes with poorly conserved components, of which only Nxt1 is identifiable in A. vaga (see “Discussion”). HP1 and KDM4 are well-conserved. Components involved in other types of histone/DNA modifications are not shown for simplicity.
To find out whether other KMTs are similarly expanded, we performed an inventory of SET domain-containing A. vaga proteins, especially those known to methylate H3K9/H3K27 (Supplementary Data 3). In addition to seven pairs of SETDB1 homologs acting on H3K9, we detected two quartets of E(z)/EZH/mes-2-like orthologs (KOG1079, Transcriptional repressor Ezh1) known to methylate H3K27. More distantly related SET-domain proteins showed domain architectures characteristic of H3K4, H3K36, and H4K20 KMTs (Trx-G/Ash1/Set1/MLL, SETD2, SETD8) and were not expanded, comprising either a quartet or a pair. Interestingly, we found six stand-alone SET-domain homology regions resembling H3K4/H3K36 KMTs (PRDM9/7/set-17), which were not predicted in the annotated gene set, non-transcribed, and lacked additional domains (KRAB_A-box, SSXRD) characteristic of PRDM9/7 proteins involved in localizing meiotic recombination hotspots and in male-specific expression54,55. Unexpectedly, we failed to identify two known KMT types acting on H3K9 or K9/K27: Su(var)3-9/SUV39H1/set-25/Clr4, a “histone read-write” architecture consisting of chromo- and SET-domains, mediating constitutive heterochromatin formation56; and G9a/EHMT2/KMT1C (ankyrin repeats plus SET), which initiates de novo methylation and silencing of repeats and developmentally regulated genes57. These domain architectures may have been lost and/or replaced by vastly expanded SETDB1-like variants.
We next sought to determine whether SETDB1 is similarly amplified in all bdelloids. Six species in the genus Rotaria from the family Philodinidae19 possess the same seven variants as do Adinetidae, indicating that SETDB1 amplification occurred prior to divergence of the major bdelloid families (Fig. 6b). An unusual SETDB1 divergence pattern is seen in the bdelloid Didymodactylos carnosus, which forms the deepest-branching sister clade to other known bdelloids51 and lacks N4CMT. While in three cases Dcar_SETDB1 forms sister clades to variants from other bdelloids, preceding quartet formation, the Q1 quartet lacks Dcar_SETDB1 homologs; moreover, an ortholog of Av_s314 shows clear evidence of loss, detected as a small 170-aa C-terminal fragment (Supplementary Data 3). This natural gene knockout is associated with an increase in LINE elements to the levels seen in monogononts, which agrees well with high concentration of 4mC over LINEs (Supplementary Fig. 16), but was not prevented by high copy number of Ago/Piwi proteins (Fig. 6c)51. Notably, LINE elements, due to their mostly vertical transmission, are expected to be more deleterious if sex is rare or absent58.
The role of MBD as a universal discriminator of 5mC marks in DNA is questioned by the presence of SETDB1 orthologs in species lacking 5mC, such as Drosophila melanogaster and Caenorhabditis elegans6, and many MBD proteins do not bind 5mC (Supplementary Fig. 14c). However, the structure of human MBD1 shows its unique potential for recognizing 5mC in the major groove without encircling DNA, which makes MBD an ideal candidate for interacting with nucleosome-bound DNA without interference from core histones59,60. Moreover, three of the seven bdelloid SETDB1-like variants display two conserved arginines in the MBD involved in the recognition of cytosines in the DNA backbone, potentially accounting for CpG preference (Supplementary Fig. 14a, b). However, they show extensive variation in the length of the antiparallel β1-β2 loop, which reaches across the major groove and interacts with one of the methyl groups. Since the overall structure is compatible with recognition of an asymmetrical DNA methyl group in the nucleosomal context, we sought to find out whether some of the seven SETDB1 variants may have adapted to preferentially recognize a novel methyl mark in the major groove.
To this end, we synthesized seven recombinant plasmids carrying tagged versions of the corresponding MBD/TAM domains (Supplementary Fig. 17a). We tested these proteins in electrophoretic mobility shift assays (EMSA) with the 451-bp repeat fragment (sAvL1-451, see above), which was either unmethylated or 4mC-methylated by N4CMT in vitro, to ensure sufficient methylation density and favorable position of methyl marks. As MBD/TAM is a generic DNA-binding domain, most AvMBD’s are capable of binding both unmethylated and methylated DNA fragments (Supplementary Fig. 17b, c). We chose AvMBD_s314 to assess its binding preference for 4mC-methylated DNA since the loss of its ortholog in D. carnosus (see above) is associated with a notable increase in LINE retrotransposon content51. We tested four AvMBD_s314 concentrations (2.38, 3.23, 3.75, and 4.14 nM) in EMSA with 32P-labeled sAvL1-451 and four concentrations of the unlabeled sAvL1-451 competitor, which was either unmethylated or 4mC-methylated by N4CMT_B in vitro. This approach provides a more adequate comparison than measurement of dissociation constants (Kd) for two labeled probes, as in vitro methylation is variably efficient. We observed a clear preference of s314 for binding 4mC-methylated DNA, with p t-test in four independent experiments (Source Data 2), when using >10× excess of non-labeled competing methylated or unmethylated DNA (p = 0.044 for 40×; p = 0.018 for 100×). Figure 6d shows a representative EMSA gel for the 3.75 nM s314 protein concentration, which yielded 88.3% protein-bound DNA with 0.05 nM 32P-labeled sAvL1-451 fragment. This protein concentration was tested twice, and the average change in the amount of unbound DNA over increasing concentrations of unlabeled competitor DNA (Fig. 6e) shows that upon the increase of competitor concentration, the shift from DNA–protein complex to unbound DNA occurs faster for 4mC-modified DNA than for unmethylated DNA, indicating a preference for 4mC target.